Der Dihydropyrimidin-Dehydrogenase-Mangel (DPD-Mangel) ist eine seltene angeborene Stoffwechselstörung mit Mangel an Dihydropyrimidin-Dehydrogenase.
Synonyme sind: Pyrimidinämie, familiäre; englisch Dihydropyrimidinuria; DPD deficiency; Familial pyrimidemia; Hereditary thymine-uraciluria
Die Erstbeschreibung stammt aus dem Jahre 1984 durch die niederländischen Kinderärzte R. Berger und Mitarbeiter.
Ursache
Der Erkrankung liegen Mutationen im DPYD-Gen auf Chromosom 1 Genort p21.3 zugrunde, welches für die Dihydropyrimidin-Dehydrogenase kodiert, die ein entscheidendes Enzym im Stoffwechsel der Pyrimidinbasen Uracil und Thymin ist.
Verbreitung
Die Häufigkeit ist nicht bekannt, die Vererbung erfolgt autosomal-rezessiv.
In Europa soll bei bis zu 9 % der Bevölkerung eine Variante am DPYD-Gen mit Verminderung der Aktivität vorliegen und bei etwa 0,5 % ein vollständiger Mangel.
Klinische Erscheinungen
Das Spektrum reicht von fehlenden Symptomen bis zu schweren neurologischen Veränderungen mit Entwicklungsverzögerung, Geistiger Behinderung und Epilepsie Hinzu können Muskelhypotonie, Mikrophthalmie, Nystagmus und Strabismus oder Autismus kommen.
5-Fluoruracil-Unverträglichkeit
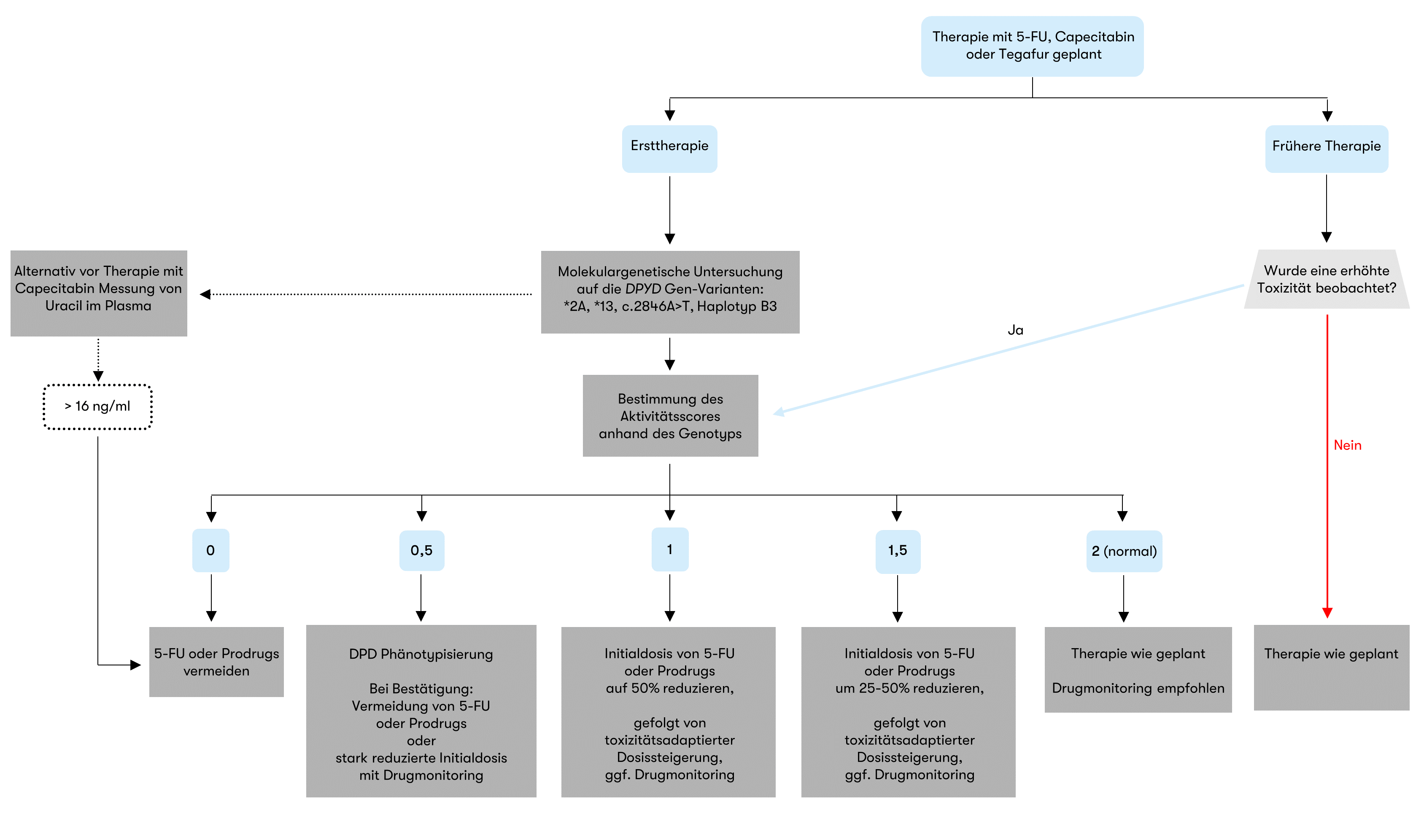
Patienten mit – unerkanntem – DPD-Mangel können unter Behandlung mit 5-Fluorouracil-haltigen Medikamenten wie Capecitabin oder Tegafur schwere toxische Nebenwirkungen erleiden. Daher soll vor einer solchen Behandlung unbedingt ein genetischer Test erfolgen.
Diagnose
Im Urin lassen sich hohe Konzentrationen von Uracil und Thymin nachweisen. Die Diagnose wird durch Humangenetik gesichert.
Therapie
Bei partiellem Mangel kann durch entsprechende Dosisanpassung eine Behandlung mit 5-FU-Medikamenten erfolgen.
Literatur
- M. de With, A. Sadlon, E. Cecchin, V. Haufroid, F. Thomas, M. Joerger, R. H. van Schaik, R. H. Mathijssen, C. R. Largiadèr: Implementation of dihydropyrimidine dehydrogenase deficiency testing in Europe. In: ESMO open. Band 8, Nummer 2, April 2023, S. 101197, doi:10.1016/j.esmoop.2023.101197, PMID 36989883, PMC 1016315 (freier Volltext).
- R. B. Diasio, T. L. Beavers, J. T. Carpenter: Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. In: The Journal of clinical investigation. Band 81, Nummer 1, Januar 1988, S. 47–51, doi:10.1172/JCI113308, PMID 3335642, PMC 442471 (freier Volltext).
- A. B. van Kuilenburg, J. Meijer, R. Meinsma, B. Pérez-Dueñas, M. Alders, Z. A. Bhuiyan, R. Artuch, R. C. Hennekam: Dihydropyrimidine Dehydrogenase Deficiency: Homozygosity for an Extremely Rare Variant in DPYD due to Uniparental Isodisomy of Chromosome 1. In: JIMD Reports. Band 45, 2019, S. 65–69, doi:10.1007/8904_2018_138, PMID 30349988, PMC 6336675 (freier Volltext).
Weblinks
- Medline Plus
- Rare Diseases
Einzelnachweise